Vorlesung: Silicatchemie
1. Einleitung
1.2. Chemische Bindung, Strukturkonzepte
1.2.1. Die Natur der Si-O-Bindung
Der Vergleich der Verbindungsbildung von Silicium und seinem leichteren
Homolgen, dem Kohlenstoff, mit Nichtmetallen zeigt, dass aufgrund der
Bindungsenergien beim Kohlenstoff C-C bzw. C-H-Bindungen, beim Silicium dagegen die Si-O-
Bindungen bevorzugt sind.
In der Strukturen der Silicate
treten als Koordinationszahlen von
Silicium meist 4, weniger
häufig auch 6 auf.
Dominierend ist die tetraedrische Koordinationssphäre, die einem sp3-Hybrid
(mit Anteilen von π-π-Bindungen zwischen O-p und Si-p sowie
sehr begrenzt auch Si-d-Zust�nden, s.u.) entspricht.
Charakteristisch für diese Koordinationszahl ist ein Abstand dSi-O von 162
pm. Seltener findet sich auch die Koordinationszahl 6, d.h. oktaederische
Umgebung (Hypervalenz wie bei SF6) mit einem typischen
Abstand dSi-O von 177 pm (Druck-Abstands-Paradoxon!). Diese Koordination
wird bei Si-O-Hochdruckphasen, bei Si-Phosphat, einigen Si-Komplexen (z.B. mit Brenzcatechin
als Ligand) und in SiF62- (d.h. mit stark elektronegativen Liganden)
beobachtet.
Aufgrund der generellen Strukturprinzipien, der Verkn�pfung von SiO4-Tetraedern
�ber unterschiedlich viele Ecken, ist Sauerstoff in Silicaten entweder von zwei
(br�ckend) oder nur von einem (endst�ndig/terminal) Si-Atom koordiniert.
Für den Bindungscharakter der Si-O-Bindung
(ionische Formulierung entsprechend Si4+ und O2- versus
kovalente Formulierung)
ergibt sich nach Pauling z.B. für Quarz ein Ionencharakter
1-e1/4(χA-χB) von 15 %.
F�r den Si-O-Abstand in Silicaten
ergibt sich
- bei Annahme einer rein ionischen Bindung aus der
Summe der Ionenradien nach Shannon ein Wert von 176 pm
(r(O2-) = 140 pm und r(Si4+) = 36 pm).
- Die berechne Si-O-Bindungslänge, die sich
bei Annahme rein kovalenter Bindung aus der Summe der Kovalenzradien
von 66 pm für O und 117 pm für Si ergeben, beträgt sogar 183 pm.
- Beobachtet wird jedoch in Silicaten ein Wert von ca. 162 pm, d.h ein geringerer Wert als nach
allen Konzepten berechnet.
Diese kurzen Bindungsabstände dSi-O
sind nur durch erheblicher Doppelbindungsanteile zu erklären (s.u.).
Innerhalb der Silicate variieren die Si-O-Abst�nde zwar nicht sehr stark, es
gibt jedoch eindeutige Tendenzen: Bereits Gibbs und Brown (1969)
haben eine Korrelation zwischen der Koordinationszahl von Sauerstoff (berechnet
mit Si und allen weiteren A-Kationen) und der Si-O-Bindungslänge
festgestellt, die in Abb. 1.2.1 dargestellt ist: Danach steigt der Si-O-Abstand
mit der Koordinationszahl von Sauerstoff, in SiO2 ist der Abstand kürzer
als z.B. in Orthosilicaten, wo die O-Atome hohe Gesamt-Koordinationszahlen aufweisen.
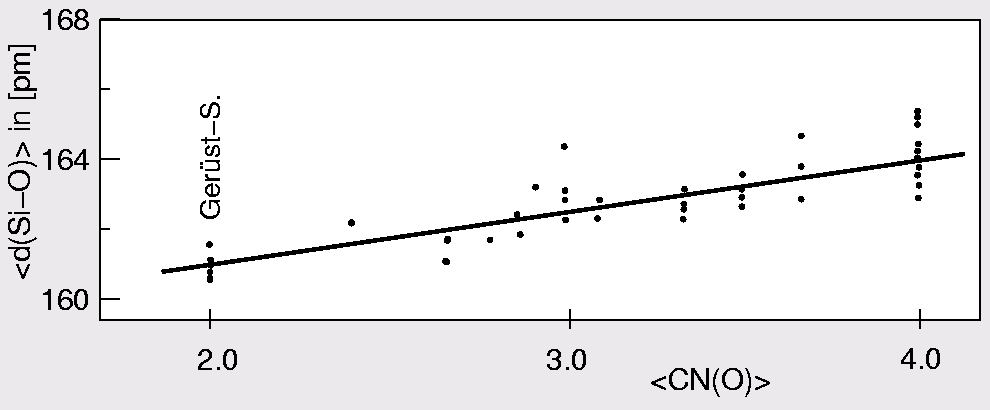 |
| Abb. 1.2.1. Korrelation des
mittleren Si-O-Abstands mit der Gesamtkoordinationszahl von Sauerstoff (nach Brown und Gibbs)
‣SVG
|
Betrachtet man lediglich die Koordination von O gegen Si, wird ebenfalls eine
klare Tendenz deutlich: Terminale (CNO=1) Si-O-Abst�nde
sind stets etwas k�rzer als Abst�nde zu verbr�ckenden Sauerstoffatomen.
Der beobachtete Bindungswinkel am Sauerstoff liegt im Durchschnitt bei 140 o,
wobei der Bereich zwischen ca. 110 und 180o schwankt. Dabei ist allerdings zu
beachten, dass die zumeist r�ntgenographisch ermittelten Si-O-Si-Winkel
in nicht wenigen F�llen durch die speziellen Lagen der Sauerstoffatome verf�lscht
sind!
Als Erklärung für diese gegenüber dem Tetraederwinkel deutlich aufgeweiteten
O-Si-O-Winkel kann wiederum der Doppelbindungsanteil der Si-O-Bindung herangezogen werden.
Die Diskussion der elektronischen Struktur von Oxido-Orthoanionen
(Orthosilicat [SiO4]4- aber auch Phosphat, Sulfat, Perchlorat usw.)
auf der Basis aktueller theoretischer Rechnungen ergibt inzwischen ein
einigermassen sicheres Bild, obwohl abh�ngig vom Rechenverfahren weiter
�ber die Anteile von π-π-Bindungen zwischen O-p und Si-p bzw. Si-d-Zust�nden
diskutiert wird (der sich auch auf die 'erlaubten' Schreibweisen f�r die Valenzstrichformeln
der Anionen auswirkt). Hierzu ist in Abbildung 1.2.2. schematisch das MO-Schema
des Orthosilicat-Ions wiedergegeben.
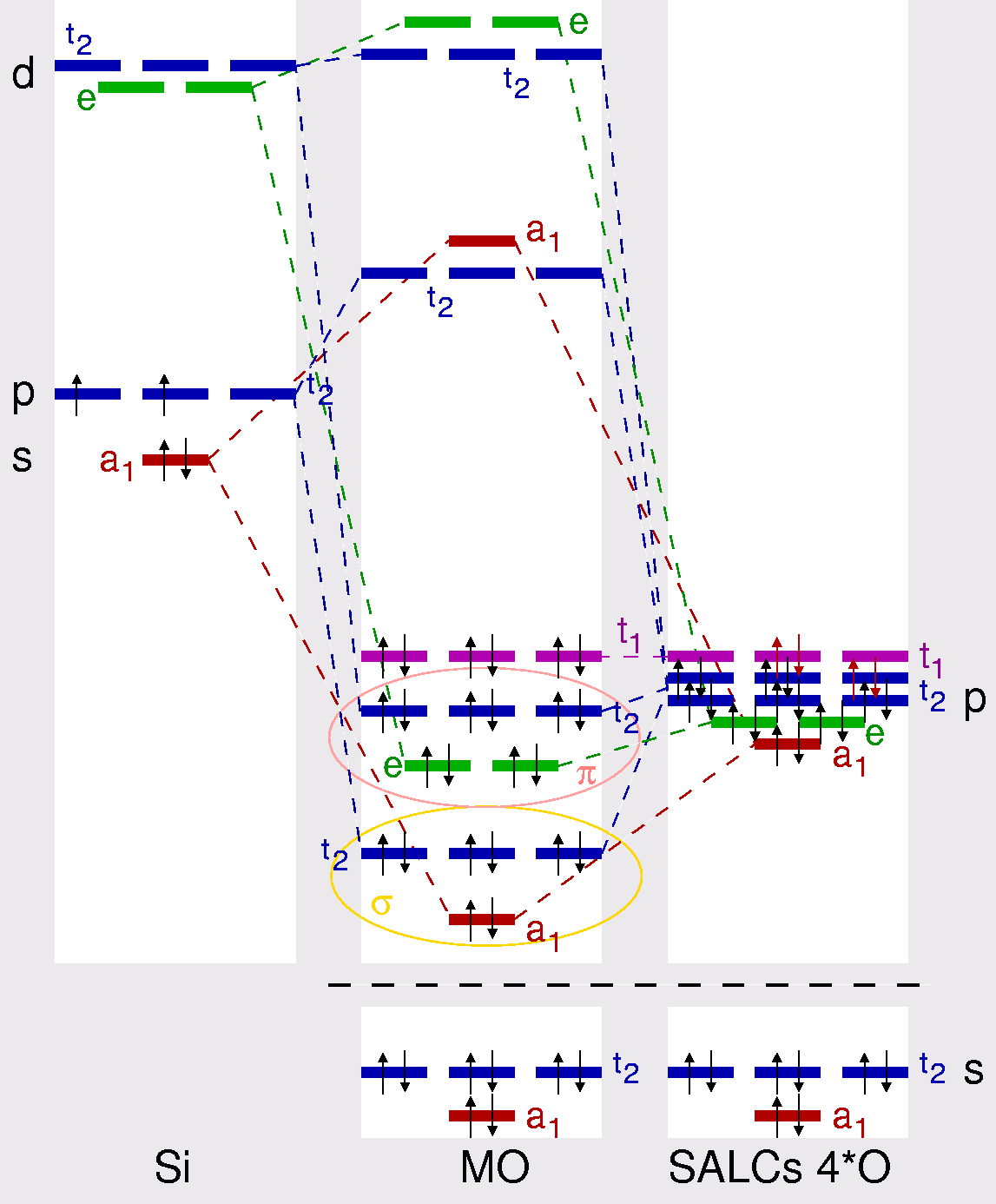 |
| Abb. 1.2.2.
Schematisches MO-Schema des Orthosilicat-Ions
‣SVG
|
Die beteiligten Atomorbitale des zentralen Si-Atoms sind Si-s (a1, in rot)
und Si-p (t2, in blau) Zust�nde, die mit vier Elektronen besetzt sind.
Die d-Orbitale bilden im Tetraeder
einen e (dz2- und dx2-y2-Orbitale, in gr�n)
und einen (t2, in blau) Orbitalsatz.
Die s-Orbitale der vier Sauerstoff-Liganden kombinieren zu
einem totalsymmetrischen a1 und einem t2-Satz. Diese
Orbitale liegen energetisch so tief, dass sie nicht an der Bindung
beteiligt sind (hohe s-p-Separation bei O). Die 12 p-Orbitale der vier O-Atome,
die mit 20 Elektronen (4+4 = 16 + 4 f�r die Ladung) besetzt sind,
bilden SALCs der irreduziblen Darstellungen a1, e, t1 und 2 x t2.
Die daraus entstehenden Molek�lorbitale des Anions sind:
- Die tiefliegenden a1- und t2-S�tze der
O-s Zust�nde sind nichtbindend (8 Elektronen).
- Die a1- und t2-Molek�lorbitale
(aus O-p und Si-s/p-AO) bilden die σ-Bindung
zwischen Si und O (8 Elektronen).
- Die e- und t2-Molek�lorbitale entstehen
durch Linearkombinationen der O-p-Zustäde (Hauptanteile)
und Si-p- (t2, in blau) bzw. Si-d-Atomorbitale (geringe Anteile).
Die Absenkung dieser beiden Molekülorbitale gegenüber den SALCs der O-Liganden rechts
representieren die π-Anteile der Si-O-Bindung.
- Der nichtbindende Orbitalsatz t1 aus O-p-SALCs
bildet den höchsten besetzten Zustand (6 Elektronen).
Selbstverst�ndlich kann ein solches lokales Molekülorbital-Modell (alleine schon
wegen der erheblichen Ladung, praktisch nicht zu berechnen!) die Bindungsverhältnisse
in einem Festkörper nicht korrekt wiedergeben.
Die Ergebnisse eine DFT-Bandstruktur-Berechnung (vollrelativitsiche FP-LAPW-Methode) von
BaSiO3 (ein synthetisches Kettensilicat)
zeigen die erheblichen Si-d-Anteile (grau unterlegt) unterhalb des Fermi-Niveaus
jedoch ebenfalls sehr deutlich (s. tDOS (oben) sowie pDOS von Si und O (unten) in Abbildung 1.2.3.).
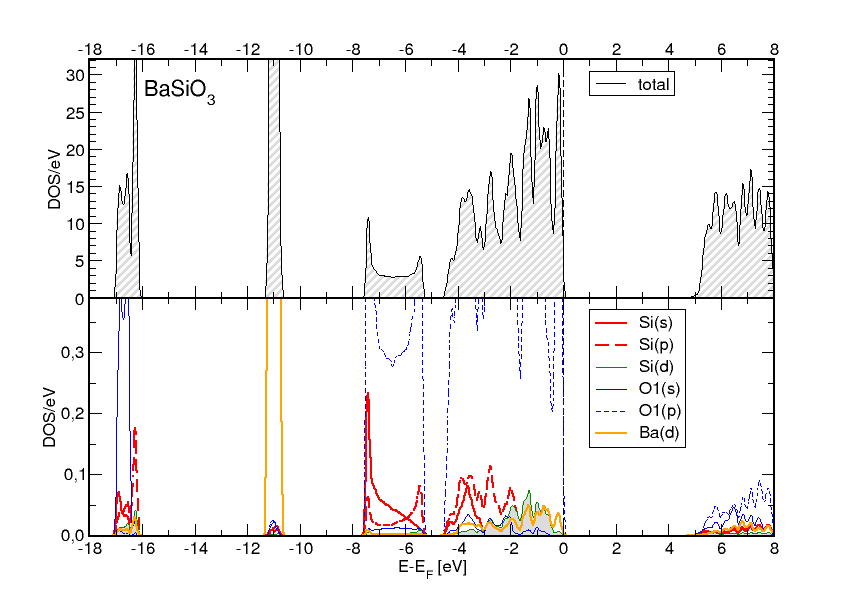 |
| Abb. 1.2.3.
Totale und partielle Zustandsdichten in BaSiO3
‣SVG
|
Die Abbildung der Elektronendichte aus dieser Rechnung (Abb. 1.2.4.) zeigt, dass
der bindungskritische Punkt (Sattelpunkt der Elektronendichte) sehr nahe
am Silicium liegt und der Wert f�r die Elektronendichte an dieser Stelle nur
sehr gering ist, der ionische Bindungsanteil ist nach dieser
Rechnung sehr deutlich.
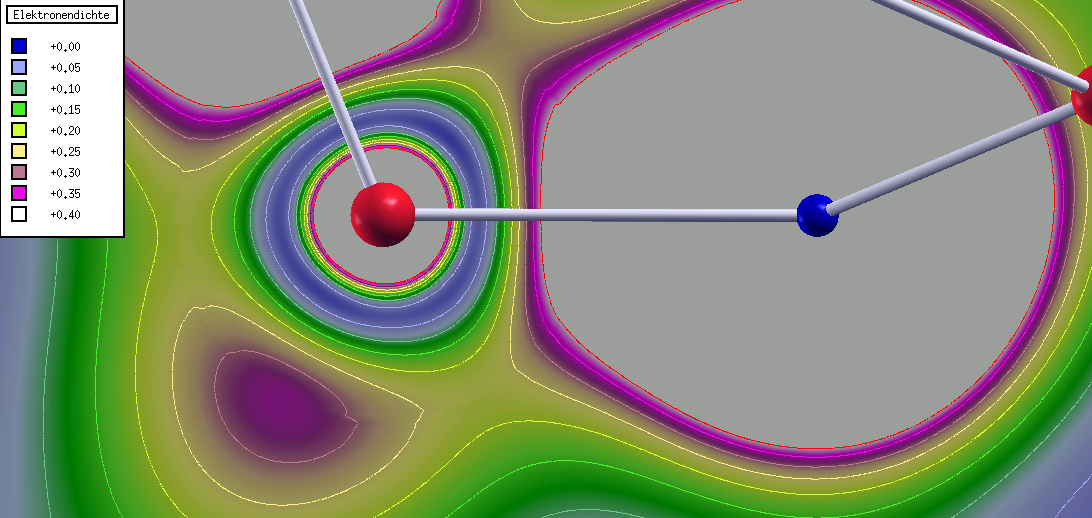 |
| Abb. 1.2.4.
Valenzelektronendichte in BaSiO3
|
1.2.2. Kationen außer Si, Wasser
Die wichtigsten Kationen in Silicaten und deren übliche Koordinationszahlen
(CN) gegenüber Sauerstoff sind in Tabelle 1.2.1 zusammengestellt.
| Kation |
CN gegen O |
| Be2+ | 4 |
| Li+ | 4, (6) |
| Al3+ | 4,6 |
| Mg2+ | (4), 6 |
| Fe2+, Ti4+ | 6 |
| Na+, Ca2+ | 6, (8) |
| K+ | 6-12 |
Tab. 1.2.1. Kationen in Silicaten
Danach können die Kationen von Be2+, Li+ und Al3+
Silicium auf den Tetraederplätzen ersetzen (sog. isomorpher Ersatz).
Aluminium kommt eine doppelte Rolle zu, es fungiert entweder als 'echtes
Kation' (Aluminium-Silicat) oder es ersetzt Silicium auf seinen Plätzen
(Alumo-Silicat). Diese Doppelrolle von Al erschwert die Interpretation
von Formeln der entsprechenden Si/Al-Verbindungen.
Die Kationen üben auf die Si-O-Bindung
einen induktiven Effekt aus, d.h. die Kationen
verstärken als Elektronen-Donatoren den Si-O-Doppelbindungscharakter.
Dadurch werden die Brückenbindungen länger (Mittelwert: 163 pm), endständige (terminale)
Si-O-Si-Bindungen werden dagegen - entsprechend einem stärkerer
Doppelbindungsanteil - kürzer (Mittelwert: 158 pm).
Die folgende empirische Regel beschreibt die entsprechende
Bindungslängenvariation:
dSi-O = 157.9 + 0.15 * CNO
Wasser ist in Silicaten fast immer als echtes Kristallwasser
eingebaut. Es sind nur wenige Hydroxy-Silicate mit [SiOx(OH)y]-Tetraedern
bekannt. Auch diese Tatsache erleichtert das Verständnis der Struktuchemie
(hydratisierter) Silicate sehr erheblich.
1.2.3. Strukturkonzepte, Pauling-Regeln
Aufgrund des relativ hohen ionischen Bindungsanteils
k�nnen die Strukturen der Silicate Ax[SiyOz]
auch h�ufig als Ionenkristalle aus O2--Anionen
und Si4+- und An+-Kationen beschrieben werden
(Konzept der Kondensation von Kationenkoordinationspolyedern, KKPs). In diesem Fall gelten
die Pauling-Regeln für Ionenkristalle, die
vereinfacht die Minimierung der potentiellen Energie in Ionenkristallen
wiedergeben. Die Pauling-Regeln gelten für Silicate fast immer.
(s. zu den Pauling-Regeln auch
Kap. 4.2. der Vorlesung
Anorganische Strukturchemie).
- Radienverhältnisregel:
Die Koordinationszahl wird in Ionenkristalle vom Verhältnis der Radien der
beteiligten Ionen bestimmt. Die Abstände selber folgen der Summe der Ionenradien.
Genauer:
Nach Einführung von Polyedern um die Kationen
(Festlegung der Koordinationszahl) und bei Zuordnung von Ionenradien zu den
einzelnen Teilchen wird die Koordinationszahl (CN) vom Radienverhältnis,
die Atomabstände vom Wert der Ionenradien bestimmt.
|
Um jedes Kation wird ein Koordinationspolyeder gebildet.
Der Abstand zwischen Kation und Anion ist durch die Summe
der Ionenradien bestimmt, die Koordinationszahl dagegen
vom Radienverhältnis.
|
Beispiele:
Bekanntestes Beispiel ist die Strukturtypenfolge (ZnS, NaCl, CsCl) bei einfachen Salzen.
Bei Silicaten ergibt sich aufgrund der Ionenradien von Si4+ und O2-
unmittelbar die Koordinationszahl 4 für Silicium gegen Sauerstoff.
Die Werte für andere Kationen entsprechen den in der Tabelle 1.2.1 oben angegebenen
und sind graphisch in Abb. 1.2.5. dargestellt.
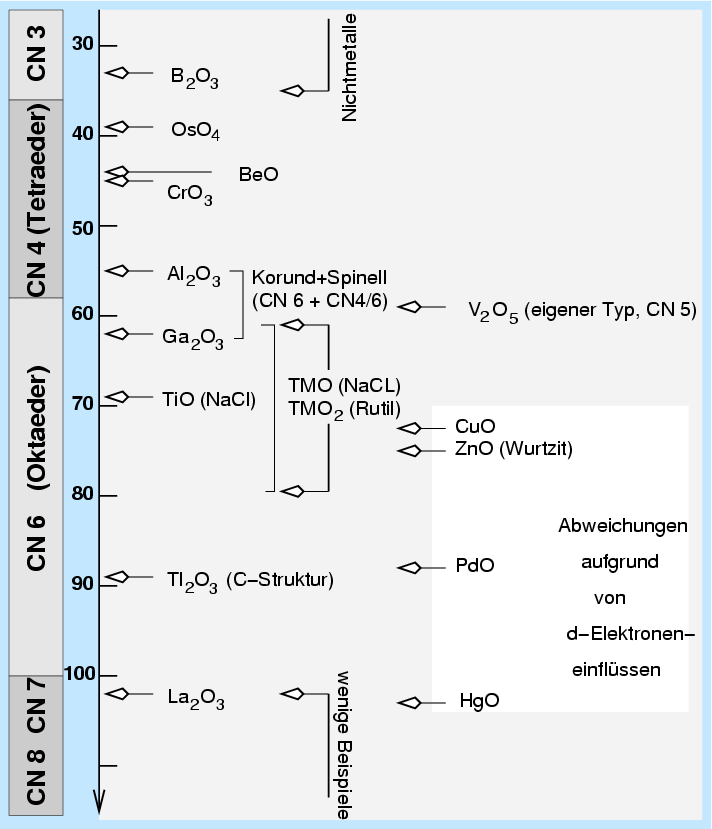 |
| Abb. 1.2.5.
Radienverhätnis-Regel und typische Kationenkoordinationen
‣SVG
|
Sind in einer Verbindung unterschiedliche Kationen vorhanden, dann
können eventuell nicht alle ideale Verhältnisse finden. In diesem Fall
weichen die Kationen mit der geringsten Ladung und dem größten Radius auf andere
Koordinationszahlen aus. Beispielsweise liegt Natrium im Sodalith
Na8[Si6Al6O24]Cl2
nicht wie sonst mit der Koordinationszahl 6, sondern nur mit einer
Koordinationszahl von 4 vor, während Si (höhere Ladung!) in CN 4
verbleibt.
- Elektrostatische Valenzsummenregel:
Für jedes Kation i mit der Ladung Z und der Koordinationszahl CN
ergibt sich die 'elektrostatische Bindungsstärke' Si zu:
Si=(Z e)/CN
Ein stabiles Ionengitter liegt dann vor,
wenn die Ladung X e der Anionen der Summe der Bindungsstärken der koordinierenden Kationen
entspricht:
X = Σi si
Die Summation erfolgt über die i-Kationen um das jeweilige Anion.
Oder als Text:
|
Die Valenz eines Anions in einer stabilen ionischen Struktur
versucht die Stärke der elektrostatischen Bindungen der umgebenden
Kationen zu kompensieren (und umgekehrt).
|
Beispiele:
- Perowskit CaTiO3:
Für die einzelen Kationen gilt:
- Ca: Z = +2; CN=12, d.h. Z/CN = 1/6
- Ti: Z = +4; CN=6 d.h. Z/CN = 2/3
Da Sauerstoff (Z = -2) von zwei Ti- und vier Ca-Atomen koordinert ist, gilt
S = 4 * 1/6 + 2 * 2/3 = 2.
D.h. also, dass die Ladung des O2- genau ausgeglichen wird.
- Spinell: MgAl2O4
Für die Kationen gilt wieder:
- Mg: Z = +2; CN=4, d.h. Z/CN = 1/2
- Al: Z = +3; CN=6, d.h. Z/CN = 1/2
Da Sauerstoff von einem Mg- und drei Aluminium-Ionen koordiniert ist, wird
auch hier die Ladung von O2- ausgeglichen ( (1+3) * 1/2 = 2)
- Granate (A32+B23+[SiO4]3):
Aufgrund der Koordinationszahlen ergeben für die Kationen (am Beispiel Grossular):
- Ca (A): Z = +2; CN=8, d.h. Z/CN = 1/4
- Al (B): Z = +3; CN=6, d.h. Z/CN = 1/2
- Si: Z = +4; CN=4, d.h. Z/CN = 1/1
Da die Oxid-Ionen von zwei Ca-, einem Al- und einem Si-Kation umgeben ist folgt
für diese Anionen:
2 * 1/4 + 1 * 1/2 + 1 * 1 = 2 (q.e.d.)
- Bei den Silicaten (weitere Rechnungen folgen bei einigen Strukturen)
gibt es maximale Abweichungen von der elektrostatischen Valenzregel von
nur 1/6.
Diese Regel kann bei Silicaten für die Bestimmung der Al-Positionen
in Alumosilicaten herangezogen werden. Ferner dient sie zur
Unterscheidung von O2-, OH- und H2O-Positionen
sowie zur Bestimmung der Oxidationszustände von Ionen (z.B. Fe2+/3+).
- 3. Pauling-Regel:
Bei Salzen mit mehreren Kationen werden diejenigen mit hoher Ladung möglichst weit voneinander weg
eingebaut,
so dass eine möglichst gute Abschirmung der Kationen voneinander möglich wird.
D.h., dass die Kationen-Koordinationspolyeder möglichst wenige Polyederelemente
gemeinsam haben sollten.
Oder als Text:
|
Teilung von Kanten und besonders von Flächen zwischen
Koordinationspolyedern reduziert die Stabilität einer Struktur.
Dieser Effekt ist besonders ausgeprägt für Kationen hoher Valenz
und geringer Koordinationszahl.
|
Beispiele:
- Bei Silicaten sind keine Beispiele für gemeinsame Flächen zwischen Tetraedern
und keine Beispiele für gemeinsame Kanten zwischen Tetraedern bekannt.
Die SiO4-Tetraeder sind stets entweder isoliert oder über gemeinsame
Ecken miteinander verknüpft.
- Der Vergleich der Reihe Silicate [SiO4]4-
-> Phosphate [PO4]3- -> Sulfate [SO4]2-
zeigt, dass wegen der in dieser Reihe steigenden Ladung des Kations
Phosphate meist nur mit wenigen gemeinsamen Ecken auftreten
(häufig isoliert, als Dimere oder maximal Ketten) und dass bei
Sulfaten keinerlei Tendenz zur Kondensation der [SO4]-Tetraeder zu
beobachten ist.
Als Erweiterung der 3. Pauling-Regel gilt, dass mehrere Kationen hoher Ladung räumlich weit voneinander
getrennt sein sollten, d.h. dass diese möglichst wenige Polyederelemente gemeinsam haben sollten.
Oder im Text
|
In einer Struktur mit mehreren Kationen weichen Kationen mit hohen Ladungen
einem Teilen von Bauelementen aus.
|
Die Konsequenz für Alumosilicate ist die sogenannte
Löwenstein-Regel, nach der niemals zwei Aluminium-Atome nebeneinander in
den Tetraederverband eingebaut sind.
- 'Sparsamkeitsregel':
Insgesamt werden möglichst wenige Koordinationen realisiert.
Im Text:
|
Die Zahl verschiedener Bauelemente in einer Kristallstruktur ist klein.
|
Allgemein ergibt sich, dass 3-dimensionale Verbände stabiler sind als zweidimensionale und
diese wiederum stabiler als isolierte Baugruppen.
Wie bei anderen Ionenkristallen auch ist h�ufig statt des Konzeptes der
KKPs auch eine Betrachtung als dichten Packungen der Anionen (Oxid-Ionen), in
der die Si- und A-Kationen die Tetraeder- und Oktaederl�cken besetzen, sehr n�tzlich.
Dies gilt insbesondere f�r Verbindungen wie z.B. die Orthosilicate, bei der
die kovalente Beschreibung �ber unterschiedlich kondensierte SiO4-Tetraeder
nicht greift.
{kind=link}
{kind=link}
{kind=link}
{kind=link}